Diagnosis of IgA nephropathy
The diagnostic feature of immunoglobulin A (IgA) nephropathy (IgAN) is the substantial accumulation of IgA in the filtering structures (glomeruli) of the kidneys. When IgAN was first described in 1968, most patients also had another antibody, IgG, in the glomeruli. The diagnosis of IgAN does not however require IgG to be detected by routine pathology studies.
Researchdone 20 years agoin Japan and Englandshowed that the IgA that accumulates in the kidneys had a unique composition: it lacked the normal amount of a specific type of sugar, galactose.This IgA is called galactose-deficient IgA(sometimes abbreviated as Gd-IgA).In the last 10 years, research done at UAB in collaboration with colleagues from around the world and supported by the IGAN Foundation of Americaand NIH has led to significant advances in our understanding of the disease. The generosity of many patients with IgA to provide blood and urine samples made these research studies possible.
Development of IgAN
Studies at UAB in collaboration with physician-scientists at other universities showed that patients with IgANoften had blood levels of galactose-deficient IgA that were higher than those in patients with other forms of kidney disease or in healthy individuals. Furthermore, most of the galactose-deficient IgA was bound by another antibody, IgG, in the blood that specifically attached to the part of the IgA that was missing the galactose sugar. The binding together of the IgA and IgG formed immune complexes (clumps) in blood, some of whichdeposit in theglomeruli. This process means that the immune system senses a part of the body to be abnormal (galactose-deficient IgA) and attacks it;thus,IgAN is an autoimmune disease and the IgG that binds the galactose-deficient IgA is called an autoantibody.
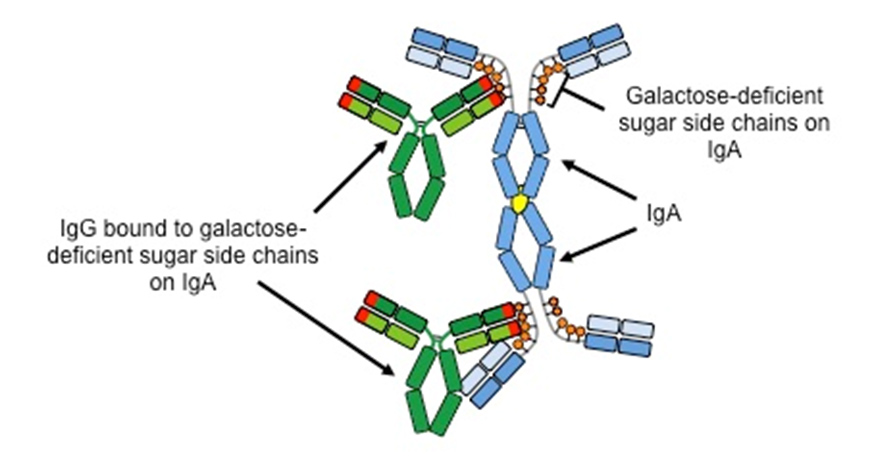
The IgA-IgG complexes in the glomeruli frequently cause structural damage that leads to leaking of red cells and protein from the blood into the urine. Persistent injury often results in scarring that reduces the filtering function of the kidneys, a condition known as chronic kidney disease. The IgA Nephropathy Research team at UAB has proposed a 4-step process as the basis for development ofIgAN.

IgG autoantibody is present in all IgAN kidney biopsies
IgG is not detected in the glomeruli in the kidney biopsy of some patients with IgAN by standard processing. This finding led some scientists to question the relevance of the 4-step process. Using recently developed laboratory tests, the UAB IgA Nephropathy Research team has shown that IgG could be detected in kidney biopsies of all patients with IgANeven when it was not seen by routine methods. Furthermore, IgG extracted from these kidney-biopsy specimens binds to galactose-deficient IgA. Using a newly available procedure with confocal microscopy, we confirmed that the IgA and IgG in the glomeruli were in overlapping positions, consistent with the two antibodies being bound together in a complex. These findings support the 4-step disease mechanism for IgANand highlight the pivotal role of IgG autoantibodies in causing the disease.
IgG is the primary autoantibody in the blood of patients with IgAN
Dr. William Placzek in the UAB Department of Biochemistry and Molecular Geneticsalong with the IgA Nephropathy Research team published a study that showedthe important role of IgG autoantibody in the formation of disease-causing immune complexes. Dr. Placzek examined the possible association between blood level of galactose-deficient IgA and blood levels of several types ofautoantibodies (antibodies that will bind to galactose-deficient IgA). The best association was found for antibodies of the IgG type. If the blood level of galactose-deficient IgA was high, the level of the IgG autoantibody was also high, and vice versa). These results suggest that IgG is the predominant type of autoantibody in IgAN.
Blood levels of galactose-deficient IgA or IgG that binds to galactose-deficient IgA (IgG autoantibody) predict long-term kidney function
The UAB IgA Nephropathy Research team partnered with investigators in China and France to discover that elevated blood levels of 1) galactose-deficient IgA and2) the corresponding IgG autoantibodiesare each higher in patients who develop chronic kidney disease 10-12 years after the diagnostic biopsy than in patients who maintained good kidney clearance function.These findings were based on work using a large group of patients. Further work is underway to refine these tests so they can be more helpful for providing prognostic information to individual patients.
Genetics of IgAN
About 5-8% of patients with IgAN have a family member with IgAN shown by biopsy, or laboratory tests suggestive of the disease (blood and/or protein inurine). Currently, there is no genetic test that can be used to reliably predict the future development of IgAN.
Studies done with investigators at Columbia University in New York have shown that the influence of several genetically determined factors increase the risk for IgAN. These studies found that variations in at least 15 areas inthe human genetic code influence the likelihood of development of IgAN. Further work has shown that some of these genetic influences affect the production of galactose-deficient IgA that can lead to higher blood levels of this immunoglobulin. Ongoing studies are trying to define the additional specific effects of these 15 genetic variations so that treatment for patients with IgAN will be focused on the root causes of the kidney injury.
New laboratory technique to measure levels of galactose-deficient IgA
Dr. Colin Reily in the Division of Nephrology at UABhas developed a better laboratory test to measure galactose-deficient IgA. This new method is an advancement that will be critical for further investigation of mechanisms involved in production of this IgA that plays a central role in the disease processes of IgAN.
Treatment of IgAN
Currently there is no disease-specific treatment of IgAN. The standard approach is to use drugs known as angiotensin converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) that can reduce the amount of protein in the urine as well as treat high blood pressure. Other measures for some patients have included fish oil supplements to reduce protein in the urine, and control of hypercholesterolemia, acidosis (too much acid in the blood based on a standard blood test), and avoidance of nicotine-containing products. With the better insight into the cause of IgAN, several pharmaceutical companies are now developing new approaches for disease-targeted treatment. Clinical trials are underway at UAB and at other medical center to evaluate new, potentially useful, medications for the treatment of IgAN.
UAB IgA Nephropathy Research Team
Dr. Jan Novak, Department of Microbiology
Drs. Bruce A. Julian, Dana V. Rizk, and Colin Reily, and Ms. CourtanyGrammer, Division of Nephrology, Department of Medicine
Drs. Matthew B. Renfrow and William J. Placzek, Department of Biochemistry and Molecular Genetics
Dr. Lea Novak, Department of Pathology
Selected publications by the IgA Nephropathy Research Team at UAB
- Berthoux F, Suzuki H, Thibaudin L, Yanagawa H, Maillard N, Mariat C, Tomino Y, Julian BA, Novak J. Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J Am Soc Nephrol 23:1579-1587, 2012.
- Berthoux F, Suzuki H, Mohey H, Maillard N, Mariat C, Novak J, Julian BA. Prognostic value of serum biomarkers of autoimmunity for recurrence of IgA nephropathy after kidney transplantation. J Am Soc Nephrol 28:1943-1950, 2017.
- Hastings MC, Moldoveanu Z, Julian BA, Novak J, Sanders JT, McGlothan KR, Gharavi AG, Wyatt RJ. Galactose-deficient IgA1 in African Americans with IgA nephropathy: serum levels and heritability. Clin J Am Soc Nephrol 5:2069-2074, 2010.
- Hastings MC, Moldoveanu Z, Suzuki H, Berthoux F, Julian BA, Sanders JT, Renfrow MB, Novak J, Wyatt RJ. Biomarkers in IgA nephropathy: relationship to pathogenetic hits. Expert Opin Med Diagn 7:615-627, 2013.
- Huang ZQ, Raska M, Stewart TJ, Reily C, King RG, Crossman DK, Crowley MR, Hargett A, Zhang Z, Suzuki H, Hall S, Wyatt RJ, Julian BA, Renfrow MB, Gharavi AG, Novak J. Somatic mutations modulate autoantibodies against galactose-deficient IgA1 in IgA nephropathy. J Am Soc Nephrol 27:3278-3284, 2016.
- Kiryluk K, Novak J. The genetics and immunobiology of IgA nephropathy. J Clin Invest 124:2325-2332, 2014.
- Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, Fasel D, Lata S, Prakash S, Shapiro S, Fischman C, Snyder HJ, Appel G, Izzi C, Viola BF, Dallera N, Del Vecchio L, Barlassina C, Salvi E, Bertinetto FE, Amoroso A, Savoldi S, Rocchietti M, Amore A, Peruzzi L, Coppo R, Salvadori M, Ravani P, Magistroni R, Ghiggeri GM, Caridi G, Bodria M, Lugani F, Allegri L, Delsante M, Maiorana M, Magnano A, Frasca G, Boer E, Boscutti G, Ponticelli C, Mignani R, Marcantoni C, Di Landro D, Santoro D, Pani A, Polci R, Feriozzi S, Chicca S, Galliani M, Gigante M, Gesualdo L, Zamboli P, Battaglia GG, Garozzo M, Maixnerová D, Tesar V, Eitner F, Rauen T, Floege J, Kovacs T, Nagy J, Mucha K, Pączek L, Zaniew M, Mizerska-Wasiak M, Roszkowska-Blaim M, Pawlaczyk K, Gale D, Barratt J, Thibaudin L, Berthoux F, Canaud G, Boland A, Metzger M, Panzer U, Suzuki H, Goto S, Narita I, Caliskan Y, Xie J, Hou P, Chen N, Zhang H, Wyatt RJ, Novak J, Julian BA, Feehally J, Stengel B, Cusi D, Lifton RP, Gharavi AG. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 46:1187-96, 2014
- Kiryluk K, Li Y, Moldoveanu Z, Suzuki H, Reily C, Hou P, Xie J, Mladkova N, Prakash S, Fischman C, Shapiro S, LeDesma RA, Bradbury D, Ionita-Laza I Eitner F, Rauen T, Maillard N, Berthoux F, Floege J, Chen N, Zhang H, Scolari F, Wyatt RJ, Julian BA, Gharavi AG, Novak J. GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway. PLoS Genet 13(2):e1006609, 2017.
- Knoppova B, Reily C, Maillard N, Rizk DV, Moldoveanu Z, Mestecky J, Raska M, Renfrow MB, Julian BA, Novak J. The origin and activities of IgA1-containing immune complexes in IgA nephropathy. Front Immunol 2016 7:117 2016.
- Lv J. Moldoveanu Z, Li Y, Kiryluk K, Gharavi AG, Novak J,Zhang H. The level of galactose-deficient IgA1 in the sera of patients with IgA nephropathy is associated with disease progression. Kidney Int 82:790-796, 2012.
- Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi AG, Fremeaux-Bacchi V, Novak J. Current understanding of the role of complement in IgA nephropathy. J Am Soc Nephrol 26:1503-1512, 2015.
- Novak J, Barratt J, Julian BA, Renfrow MB. Aberrant glycosylation of the IgA1 molecule in IgA nephropathy. SeminNephrol 38:461-476, 2018.
- Placzek WJ, Yanagawa H, Makita Y, Renfrow MB, Julia, BA, Rizk DV, Suzuki Y, Novak J, Suzuki H. Serum galactose-deficient-IgA1 and IgG autoantibodies correlate in patients with IgA nephropathy. PLoS One 13:e0190967, 2018.
- Reily C, Rizk DV, Julian BA, Novak J. Assay for galactose-deficient IgA1 enables mechanistic studies with primary cells from IgA nephropathy patients. Biotechniques 65:71-77, 2018.
- Rizk DV, Maillard N, Julian BA, Knoppova B, Green TJ, Novak J, Wyatt RJ. The emerging role of complement proteins as a target for therapy of IgA nephropathy. Front Immunol10:504, 2019.
- Rizk DV, Saha MK, Hall S, Novak L, Brown R, Huang ZQ, Fatima H, Julian BA, Novak J. Glomerular immunodeposits of patients with IgA nephropathy are enriched for IgG autoantibodies specific for galactose-deficient IgA1. J Am Soc Nephrol 30:2017-2026, 2019.
- Suzuki H, Fan R, Zhang Z, Brown R, Hall S, Julian BA, Chatham WW, Suzuki Y, Wyatt RJ, Moldoveanu Z, Lee JY, Robinson J, Tomana M, Tomino Y, Mestecky J, Novak J. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest 119:1668-77, 2009.
- Suzuki H, Krzysztof K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, Wyatt RJ, Scolari F, Mestecky J, Gharavi AG Julian BA. The pathophysiology of IgA nephropathy. J Am Soc Nephrol 10:1795-803, 2011.
- Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med 368:2402-14, 2013.